Zajednička karakteristika ovih bolesti je rasprostranjena ćelijska infiltracija i nagomilavanje ekstraćelijskog matriksa distalno od terminalnih bronhiola, u području alveolarnih zidova i međualveolarnog prostora - u intersticijumu pluća.
Difuzne bolesti plućnog parenhima tj. intersticijske bolesti pluća nastaju kao rezultat različitih poremećaja izazvanih hroničnim oštećenjima plućnog parenhima i obuhvataju kako bolesti poznatog uzroka (na pr. kolagene vaskularne bolesti, bolesti koje su u vezi sa životnim okruženjem ili sa lekovima i dr.) tako i bolesti nepoznatog uzroka (idiopatske intersticijumske pneumonije (IIP), granulomatozne bolesti pluća i drugi oblici intersticijumskih bolesti pluća (IBP) uključujući limfangiolejomiomatozu (LAM), histiocitozu X (HX) i eozinofilnu pneumoniju...
Sistemska klasifikacija na osnovu etiologije ili specifičnih obeležja oboljenja je najjednostavnija, na IBP poznate I IBP nepoznate etiologije. IBP nepoznate etiologije čine 65% od svih IBP.

Slika Etiološka klasifikacija intersticijskih plućnih bolesti
Difuzne intersticijumske bolesti pluća se, prema Američkom Torakalnom Udruženju, dele na 4 grupe: (1) difuzne intersticijske bolesti pluća poznate etiologije ili povezanosti kao što su one izazvane ekspozicijom organskim (hipersenzitivni pneumonitis-HP) ili neorganskim materijama (azbestoza), lekovima, zračenjem, ili povezane sa kolageno-vaskularnim bolestima, kao što su sistemska skleroza i reumatoidni artritis, (2) idiopatske intersticijske pneumonije, (3) granulomatoze i (4) retke parenhimske bolesti sa jasno definisanim kliničko-patološkim nalazom (LAM-limfangiolejomiomatoza, plućna histiocitoza Langerhansovih ćelija, eozinofilna pneumonija itd).
Intersticijske bolesti pluća imaju jasne zajedničke karakteristike:
- tipične histološke karakteristike
- komparabilne RTG / CT nalaze,
- tipične poremećaje u plućnoj funkciji tj. plućnoj fiziologiji,
- sličnost simptomatologije
Zajednička karakteristika ovih bolesti je rasprostranjena ćelijska infiltracija i nagomilavanje ekstraćelijskog matriksa distalno od terminalnih bronhiola, u području alveolarnih zidova i međualveolarnog prostora - u intersticijumu pluća.
Idiopatska plućna fibroza (IPF)
Idiopatska plućna fibroza (IPF) je definisana kao specifičan oblik hronične, progresivne, fibrozirajuće intersticijumske pneumonije nepoznatog uzroka, koja se dešava primarno kod odraslih osoba, a koja je pritom ograničena na pluća i udružena sa histopatološkim i/ili radiološkim nalazom UIP (Uobičajena tj. obična intersticijska pneumonija).
Kliničke karakteristike. Postepeno progredirajuća dispnea koja traje duže od 3 meseca, suv kašalj (paroksizmalan, refrakteran na antitusike) uz auskultacijski nalaz kasno inspirijumskih pukota nad plućnim bazama je karakteristika kliničkog ispoljavanja ove bolesti u početnoj fazi. Ovakvi simptomi u odsustvu spirometrijskih poremećaja i kod normalnog nalaza radiografije pluća, uz isključenje srčane dekompenzacije i još nekih bolesti drugih organa koja mogu biti uzrok ovakvih nalaza (bubrežna insuficijencija...) treba da upute lekara na sumnju da se možda radi o intersticijskoj bolesti pluća tipa IPF.
Od IPF najčeše obolevaju osobe između 40. i 70. godine života. Bolest se češće javlja kod muškaraca. Oboleli se najčešće javljaju lekaru zbog postepeno progredirajuće dispnee, koja je kod većine prisutna duže od 6 meseci pre prvog pregleda. Dispneju često prati suv kašalj i osećaj stezanja u grudima. Kašalj može biti paroksizmalan i često je refrakteran na antitusike. Opšte tegobe su često prisutne od samog početka: gubitak telesne mase, malaksalost, osećaj umora, ali se mogu javiti i tokom napredovanja bolesti. Vanplućne manifestacije bolesti nisu karakteristične za ovo obolenje, što je od značaja za diferencijalnu dijagnozu.
Auskultacijom nad plućnim bazama, može se kod 80 % bolesnika u početnom stadijumu bolesti utvrditi prisustvo kasno inspirijumskih pukota. Sa napredovanjem bolesti pozitivan auskultatorni nalaz se proširuje prema vrhovima pluća. Kasnije tokom bolesti dolazi i do promene karaktera pukota (pridružena infekcija, kardijalna dekompenzacija...). Batičasti/maljičasti prsti se razvijaju u do 70% obolelih. Maljičasti prsti se često javljaju i kod bolesnika sa difuznom plućnom fibrozom izazvanom reumatoidnim artrtitisom i azbestozom, ali se ne javljaju kod drugih benignih difuznih intersticijalnih fibroza (sarkoidoze, hipersenzitivnog pneumonitisa, histiocitoze, limfangioleiomio-matoze). Prisustvo maljičastih prstiju znači povećava verovatnoću da je difuzni plućni proces zaista IPF, ali njihovo odsustvo nije od većeg dijagnostičkog značaja.
Centralna cijanoza često udružena sa teškom tahipneom i opterećenje desnog srca su neke od manifestacija veoma uznapredovale bolesti.
Kako bolest napreduje, javlja se centralna cijanoza koja je često povezana sa teškom tahipneom. Bolesnici sa IPF hiperventiliraju da bi kompenzovali hipoksiju. Pothranjenost se viđa kod bolesnika u uznapredovaloj fazi bolesti sa insuficijencijom desnog srca. Kako napreduje obliteracija plućnog krvotoka fibroznim procesima, povećava se plućna hipertenzija. U preterminalnoj fazi bolesti, teška hipoksija može dovesti do kardijalne dekompenzacije, uzrokujući kliničke manifestacije insuficijencije levog srca ili aritmije.
Najčešći uzrok smrti je respiratorna insuficijencija, potom srčana insuficijencija, ishemijska srčana bolest, infekcija i plućna embolija. Učestalost bronhogenog karcinoma je povećana (10% do 15% bolesnika) kod bolesnika sa uznapredovalom IPF. Prognoza pacijenata sa udrženim prisustvom IPF i karcinoma bronha je veoma loša.
Smrtnost od IPF raste sa životnom dobi. Brojnim studijama je utvrđeno da se srednja dužina preživljavanja od momenta postavljanja dijagnoze kreće između 2 i 5 godina.
Dijagnostički pristup kod IPF obuhvata sledeće postupke: anamnezu i fizički pregled kao prvi korak, laboratorijske i serološke testove, testove ispitivanja plućne funkcije, radiološka ispitivanja (primena standardizovane radiografije i HRCT tj. visokorezolutivne kompjuterizovane tomografije) i biopsiju plućnog tkiva sa histološkim pregledom bioptičkog materijala (slika)
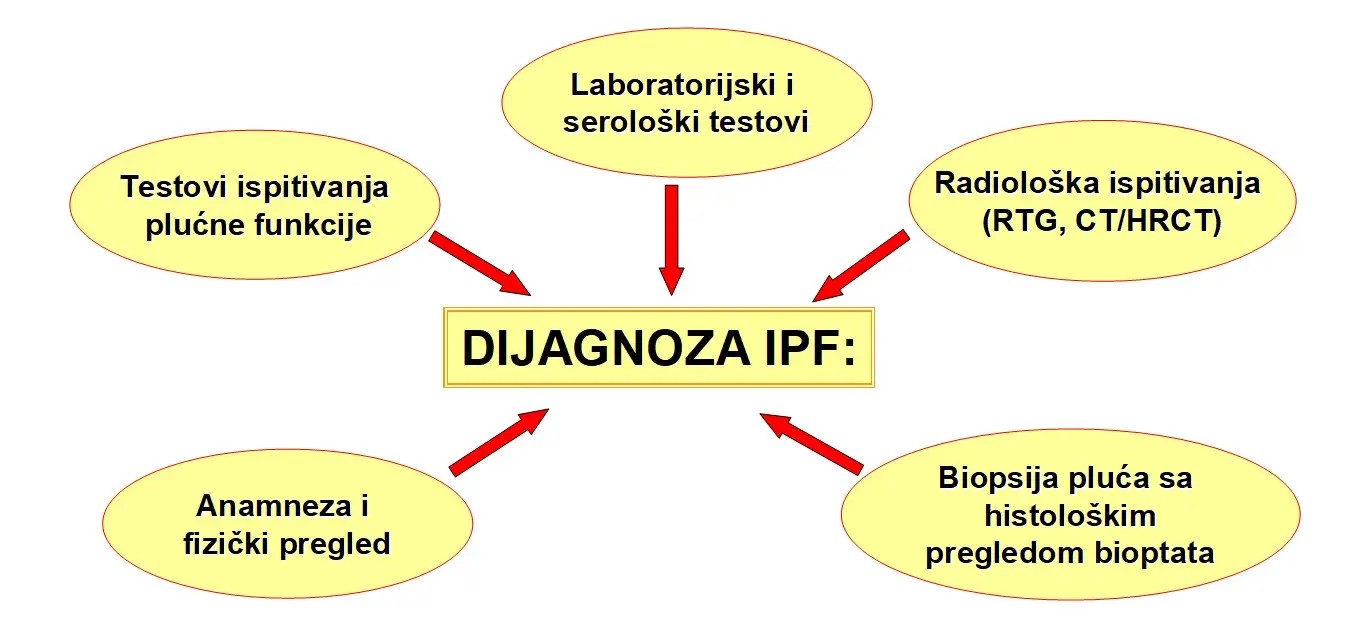
Slika . Dijagnostički algoritam idiopatske plućne fibroze
Postavljanje sumnje lekara PZZ na osnovu anamneze I fizikalnog nalaza za ovu bolest je od presudnog značaja za ranu dijagnostiku ovog obolenja, s obzirom na snižen difuzijski kapacitet i druge parametre difuzije u samom početku bolesti čak kao jedini patološki nalaz, a koji se mogu utvrditi samo u tercijernim zdravstvenim ustanovama koje imaju tehničke uslove za ispitivanje plućne funkcije.
Suština dijagnostičkog algoritma IPF je da se isključe druga, slična intersticijska obolenja, kako bi se multidisciplinarno postavila dijagnoza IPF i definisao terapijski pristup.
Tačnost dijagnoze IPF raste sa kliničkom, radiološkom i histopatološkom korelacijom - najvažniji je multidisciplinarni konsenzus. Dijagnoza IPF se zasniva na kliničkoj slici, specifičnoj kombinaciji nalaza HRCT i hiruške biopsije ukoliko je radjena.
Testovi plućne funkcije ukazuju na snižen difuzijski kapacitet u samom početku bolesti čak kao jedini patološki nalaz. Sa daljim razvojem bolesti i fibroze, registruje se restrikcijska insuficijencija ventilacije sa hipoksemijom, kasnije i hiperkapnijom. Testovi ispitivanja plućne funkcije se koriste i za praćenje toka bolesti (njene progresije ili dobrog odgovora na terapiju). Gasne analize arterijske krvi pri opterećenju su senzitivan parametar za praćenje kliničkog toka bolesti.
Kompjuterizovana tomografija visoke rezolucije (HRCT). Ima veliki značaj u dijagnostici intersticijumskih bolesti pluća zbog toga što je mnogo senzitivnija i mnogo specifičnija metoda od standarne radiografije grudnog koša. Primarna uloga HRCT je da se izdiferenciraju pacijenti sa nalazom tipičnim za UIP/IPF od nalaza kod ostalih difuznih intersticijumskih lezija. Primena HRCT omogućava kliničaru uvid u proširenost i težinu lezija u plućnom parenhimu. Korisna je i za dijagnostiku i procenu "aktivnosti" bolesti - omogućava kliničaru uvid u proširenost i težinu lezija.
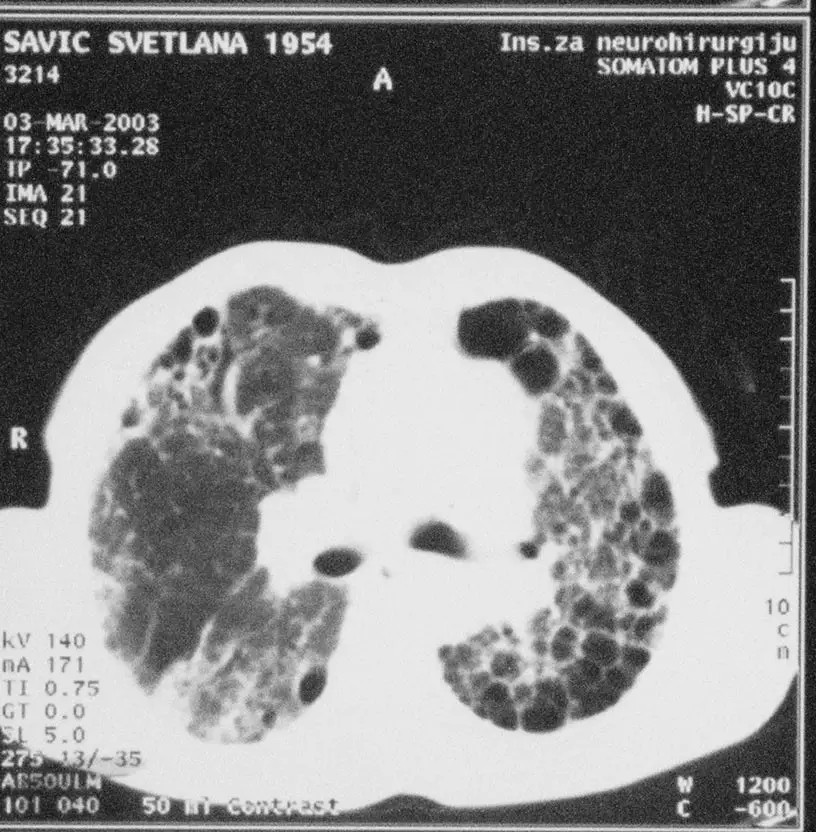
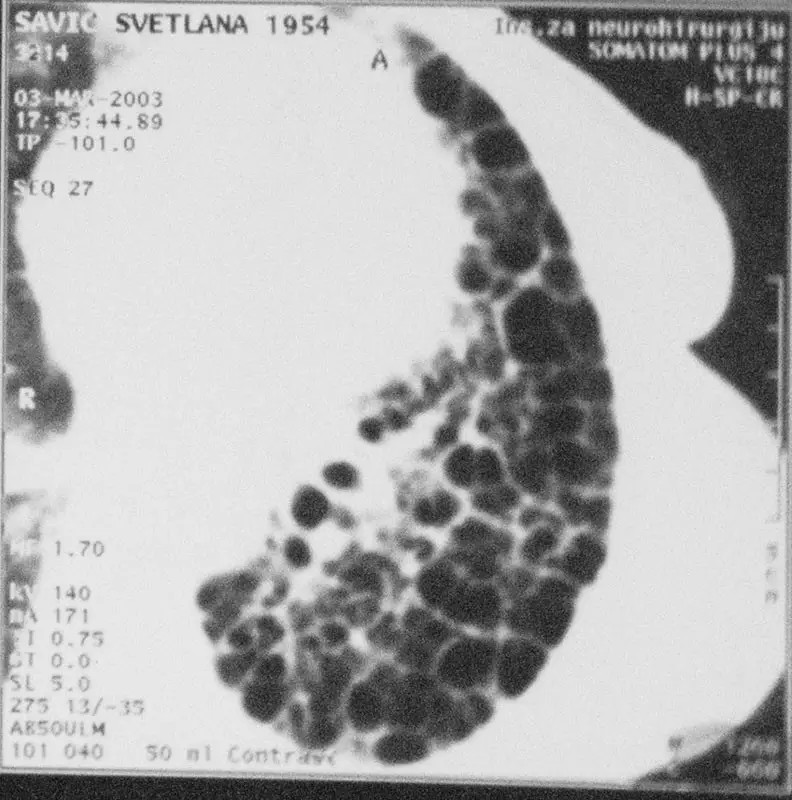
Sl…HRCT kriterijumi za dijagnozu UIP, saćasto pluće
Terapija IPF podrazumeva primenu antifibrotske terapije, dostupna su 2 leka, Pirfenidon (ESBRIET) i Nintedanib (OFEV).
Kriterijumi za procenu težine intersticijskih promena obuhvataju:
- Procenu dispnoje
- Rezultate testova plućne funkcije
- Kardiopulmonalni test opterećenja (uključujući
- gasne analize u mirovanju i pri naporu)
- Kompjuterizovanu tomografiju visoke rezolucije (HRCT)
- PH nalaz biopsije ako je rađena?
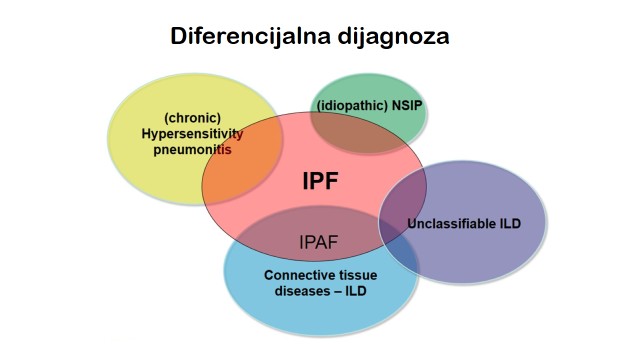
Diferencijalna dijagnostika - na osnovu kliničkih pokazatelja
Za pravilno postavljanje dijagnoze IPF od presudnog je značaja dobro uzeta anamneza. Naročito je značajno uzeti detaljnu anamnezu radne i životne sredine. Azbestoza i izlaganje teškim metalima, mogu dati identičnu kliničku sliku kao i IPF. Hronična fibrozirajuća forma hipersenzitivnog pneumonitisa uzrokovana produženim izlaganjem antigenu, može klinički takođe odgovarati IPF. Plućna bolest može biti dominantna kod brojnih kolageno-vaskularnih bolesti, tako da tek pažljivo uzeta anamneza može da ukaže na prisustvo Raynaud-ovog fenomena, artritisa, miozitisa, „sicca” sindroma i gastroezofagealne regurgitacije i da našu dijagnostiku navede na pravi put. Moraju se detaljno uzeti podaci o lekovima koji se koriste, da bi se isključile difuzne plućne bolesti njima izazvane (citotoksični agensi, nitrofurantoin, amiodaron itd.). Od velikog značaja je i prethodno ili aktuelno prisustvo maligne bolesti, pošto karcinomatozni limfangitis može imati iste karakteristike kao i fibroza kod IPF.
Fibrozirajući alveolitis povezan sa kolageno-vaskularnim bolestima, razlikuje se od IPF po tome što je prisustvo i težina respiratornih simptoma kod prvih vrlo nestalna i nije u korelaciji sa masivnošću intersticijalnih plućnih promena utvrđenim testovima plućne funkcije ili HRCT-om). Plućna bolest je često već jako uznapredovala u momentu otkrivanja, što je često posledica ograničene pokretljivosti bolesnika zbog muskulo-skeletnih promena. S druge pak strane, prisustvo sistemske bolesti može uputiti kliničara na mogućnost plućne lokalizacije bolesti, te se time može fibrozirajući alveolitis dijagnostikovati u svojoj ranoj, asimptomatičnoj fazi, često na rutinskom, radiografskom staging-u.
U pojedinim slučajevima mogu se javiti klinički pokazatelji koji ukazuju na to da je početna dijagnoza IPF netačna. Prisustvo kožnih promena kao što je lupus pernio ili nodozni eritem može ukazati na sarkoidozu. Detaljan fizikalni pregled može takođe ukazati na prisustvo reumatoidne zglobne bolesti, a sklerodaktilija u kombinaciji sa plućnom fibrozom je dijagnostički kriterijum sistemske skleroze.
Dob bolesnika koji prvi put dolazi na pregled može biti od presudnog značaja za postavljanje prave dijagnoze. IPF je bolest starije životne dobi i javlja se uglavnom kod osoba starijih od 50 godina, dok se na pr. sarkoidoza javlja kod mlađih i sredovečnih osoba. Histiocitoza se javlja kod mladih pušača, dok se limfangioleiomiomatoza javlja uglavnom kod žena u predmenopauzi.
Praćenje kliničkog toka IPF
Iako je IPF progresivna intersticijalna plućna bolest, intenzitet i stepen progresije značajno variraju od bolesnika do bolesnika, tako da trenutno nije moguće izdvojiti ni kliničke ni patohistološke stadijume IPF.
Objektivno, validnost parametara za procenu aktivnosti bolesti je različita od studije do studije. Na žalost, samo mali broj obolelih od IPF reaguje na terapiju. Subjektivno poboljšanje je često (i do 70% lečenih bolesnika) i ne treba da se koristi kao jedini faktor za procenu nastavka terapije. Objektivno poboljšanje parametara plućne funkcije se javlja samo u 20-30% lečenih.
Podaci o toku bolesti u većini slučajeva IPF su ograničeni. U stvari, većina objavljenih studija ukazuje na to da se oboleli od IPF u najvećem broju slučajeva javljaju lekaru u već uznapredovaloj fazi bolesti. Ovaj podatak je veoma značajan, pošto opisi većine slučajeva koji su udruženi sa progresijom bolesti mogu biti relevantni samo za terminalnu fazu bolesti. Ovakvi nalazi ukazuju na to da je potrebno uložiti dodatne napore da bi se bolesnici sa IPF otkrivali u ranijoj fazi bolesti.
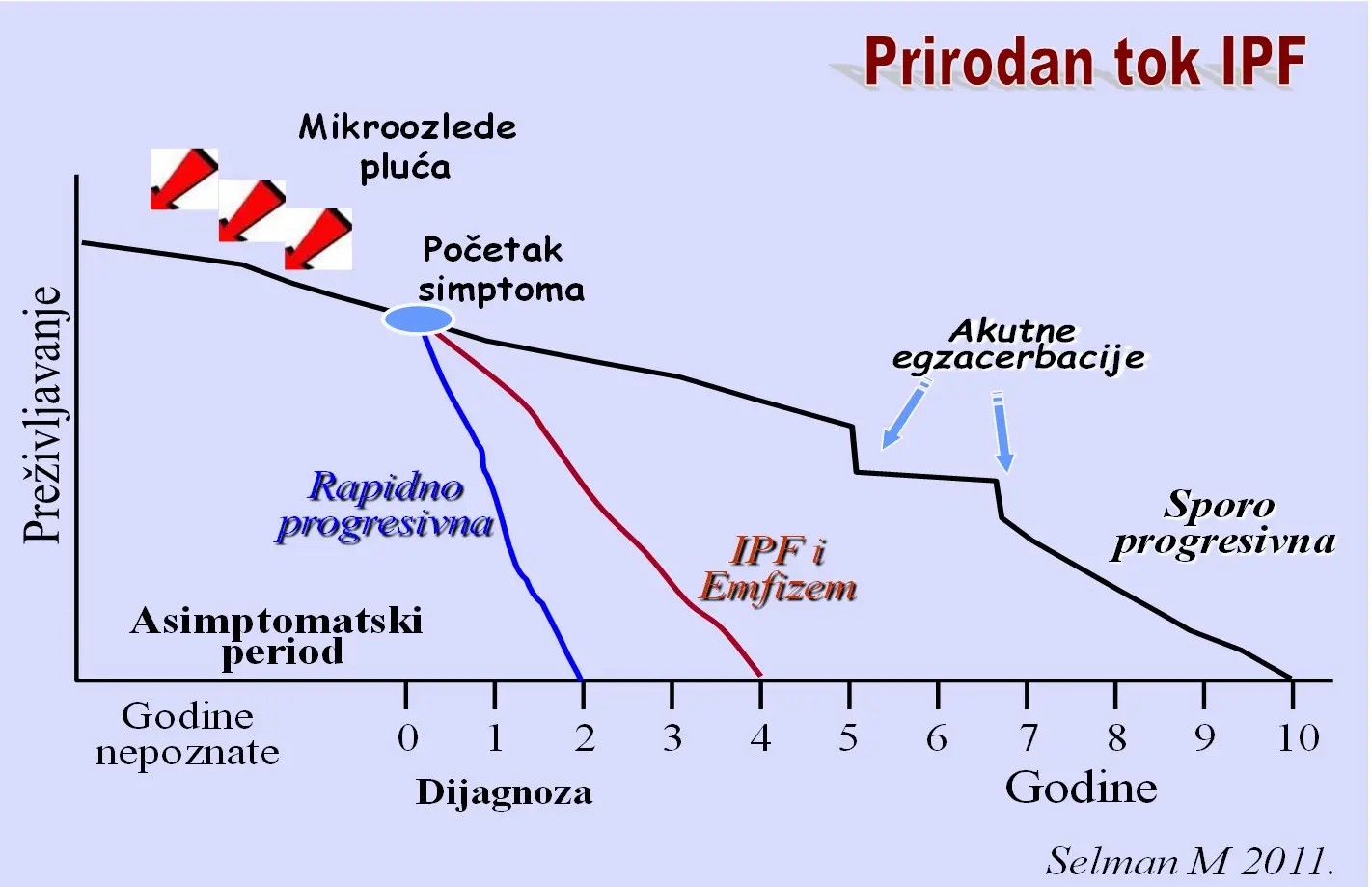
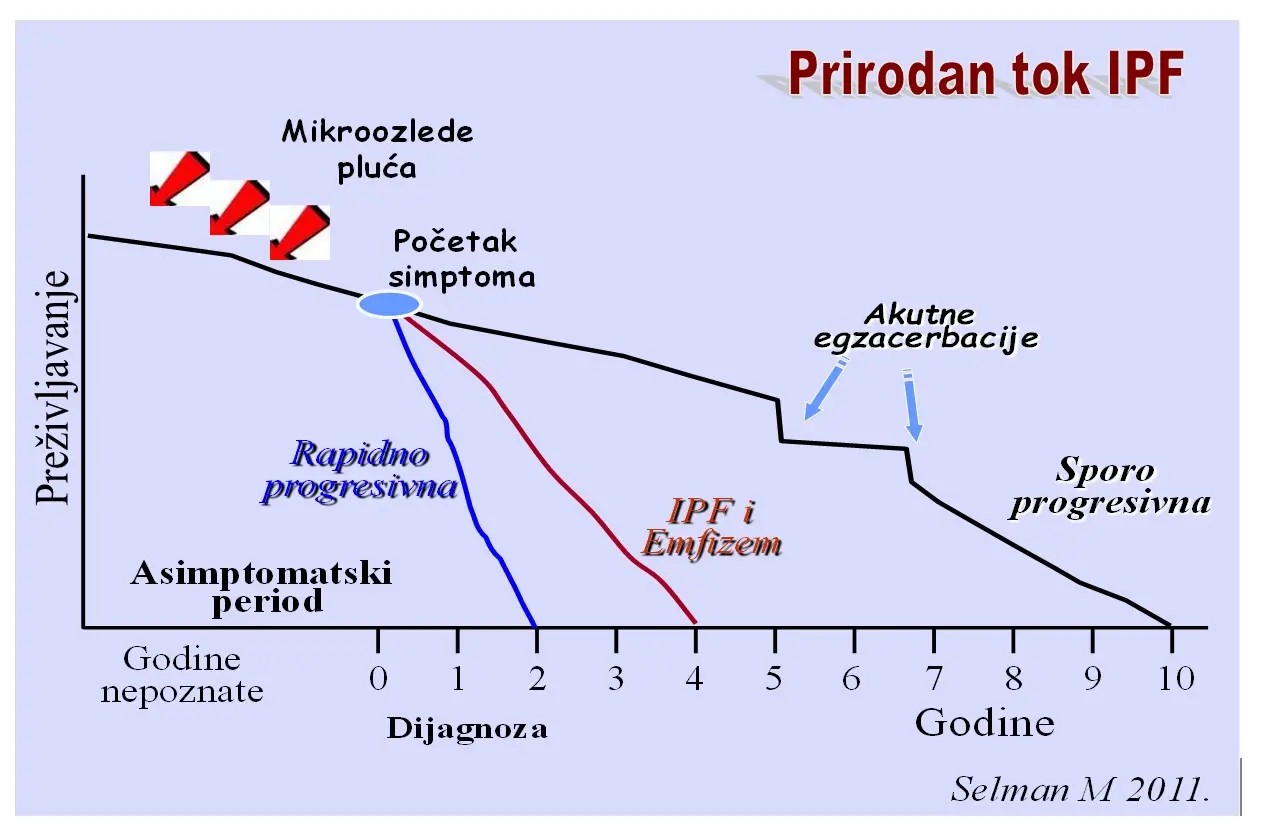
Slika Varijante prirodnog toka IPF
Kliničke varijante manifestacija u IPF:
- Stabilna ili sporo progresivna bolest
- Rapidni progresori (akcelerirana varijanta)
- “Idiopatska” Akutna pogoršanja (egzacerbacije) tj. AE-IPF
- Kombinacija IPF i Emfizema (CPFE)
AKUTNO POGORŠANJE IPF - Kriterijumi za akutno pogoršanje IPF su neobjašnjivo pogoršanje dispnee u periodu od mesec dana, uzrok pogoršanja ne može biti identifikovan (npr. pneumonija, plućni embolizam, pneumotoraks, srčana slabost), hipoksemija (pogoršanje ili teško oštećenje gasne razmene), pojava novih infiltrata na radiografiji pluća. Histološki: kao akutno ili organizirajuće difuzno alveolarno oštećenje (DAD) ili organizirajuća pneumonija (ređe).
Kombinacija Plućne Fibroze i emfizema - CPFE
CPFE-poseban IPF fenotip povlači značajno veći mortalitet od IPF
Prirodan tok bolesti i prognoza
IPF je fatalna bolest. Oko 50% umire unutar 3-5 godina.
Prirodan tok i prognoza bolesti su varijabilni i nepredvidivi:
a.Najveći broj pac. sa IPF razvija postepeno pogoršanje plućne funkcije tokom godina. Manjina ostaje stabilna ili se pogoršava rapidno.
b. Neki pac. imaju epizode akutnog respiratornog pogoršanja uprkos prethodnoj stabilnosti bolesti
- Progresija bolesti se manifestuje povećanjem respiratornih simptoma, pogoršanjem nalaza testova plućne funkcije, progresivnom fibrozom na HRCT-u, akutnim respiratornim pogoršanjem, ili smrtnim ishodom.
- Pacijenti sa IPF mogu imati supklinička ili ispoljena komorbidna stanja uključujući plućnu hipertenziju, gastroezofagealni reflux, opstruktivnu sleep apneu, obesitas i emfizem.
- Impakt ovih stanja na ishod u pacijenta sa IPF, nije potpuno jasan.
Činioci povišenog rizika od smrti kod IPF mogu se podeliti u grupu faktora rizika od smrti u vreme dijagnoze, tj. početne faktore, i u grupu tzv. longitudinalnih faktora rizika od smrti koji odražavaju dinamiku bolesti u vremenu.
- Početni faktori
- Nivo dispnee
- DLco < 40%
- SaO2 ≤ 88% tokom 6 MWT
- Obim saćastih promena na HRCT
- Plućna hipertenzija
- Longitudinalni faktori
- Pogoršanje dispnee
- Sniženje FVC ≥ 10% od apsolutne vrednosti
- Sniženje DLco ≥ 15% od apsolutne vrednosti
- Pogoršanje fibroze na HRCT
Uzroci smrti u IPF.
Bolesnici sa IPF umiru najčešće zbog respiratorne insuficijencije (35-40%), kardiovaskularne bolesti sa srčanom insuficijencijom (oko 30%), a zatim u mnogo manjem procentu - oko 20%, od ostalih uzroka, u desetak procenata od nadovezanog karcinoma pluća i dr.
Terapija IPF
Tendencija savremenih vodiča za terapiju IPF, ATS/ERS/JRS/ALAT 2015. Podrazumeva primenu antifibrotske terapije, dostupna su 2 leka, Pirfenidon (ESBRIET) i Nintedanib (OFEV).


